 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipThere are several strategies employed when a peptide synthesis requires optimization. Typically, the first thing considered is whether or not to double couple specific amino acids within the sequence. This is somewhat of a change in mentality from traditional room temperature synthesis strategies where double coupling is frequently used for the entire peptide sequence.
In a previous post, I briefly described several scenarios in which doubling coupling can be used in conjunction with microwave heating to improve the overall crude peptide purity. In today’s post, I will delve more deeply into the question of whether or not double coupling is necessary to improve your peptide synthesis.
The most common circumstance in which double coupling an amino acid is considered often involves arginine. This amino acid, with its guanidine-containing side chain accompanied by the Pbf protecting group is one of the largest, most sterically hindered, amino acids. Coupling Arg residues has been considered historically challenging due to the extreme steric bulk, despite containing a slightly more reactive amino nitrogen (pKa = 9.04).
To further explore the impact of steric bulk and how double coupling may be used to overcome this challenge, I decided to synthesize octa-Arg, a commonly synthesized peptide that has the potential to deliver peptide– and non-peptide-based cargos to a cell’s interior.
I first synthesized octa-Arg using my Biotage® Initiator+ Alstra™ on ChemMatrix® Rink amide resin with DIC and Oxyma as my coupling reagents. For this first synthesis I used single couplings which proceeded at 75°C for 5 minutes. I wanted to see how bad the product can really be. To my surprise, the desired peptide was present with few impurities, Figure 1.
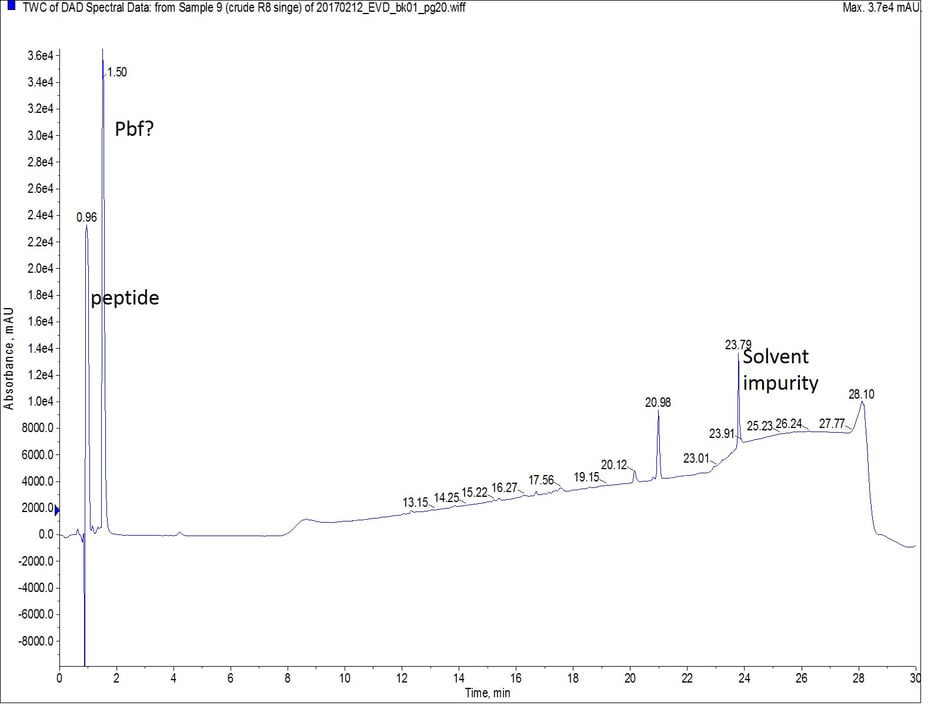
Figure 1: Crude analytical UV HPLC spectrum of octa-Arg synthesized with single couplings. There is no evidence of the peptide eluting later in the gradient.
As octa-Arg is highly charged, especially under standard reversed phase conditions, achieving any degree of column retention by the C18 stationary phase was very difficult. The mass spectrum of the first UV peak though, clearly shows the desired peptide, along with incremental TFA ion pairs as expected, Figure 2. I’ll save optimizing this chromatography for a more thorough discussion later though.
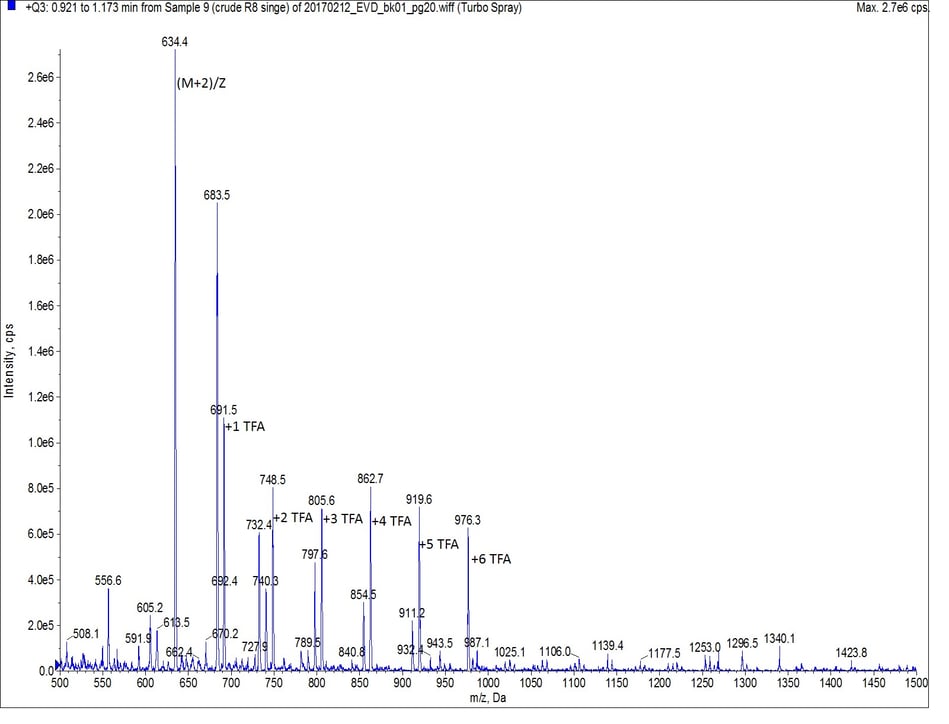
Figure 2: Mass spectrum of the main peptide peak (r.t. 0.96 min). The peptide is evident by the doubly charged species with MW = 634.5 Da. All other labeled peaks represent the TFA ion pairs of the doubly charged peptide. Each peak is labeled with the number of TFA ions paired.
For the second synthesis of octa-Arg, I employed a different strategy. Keeping the coupling reagents, scale, and equivalents constant, I repeated the synthesis with single couplings only this time I reduced the coupling temperature to 50°C and extended the reaction time to 20 minutes. Extending the coupling time is a strategy that is sometimes used for difficult couplings when using room temperature synthesis, and one that I have also used for microwave-assisted peptide synthesis when reagent cost is a forefront concern. This approach too yielded a successful synthesis, Figure 3.
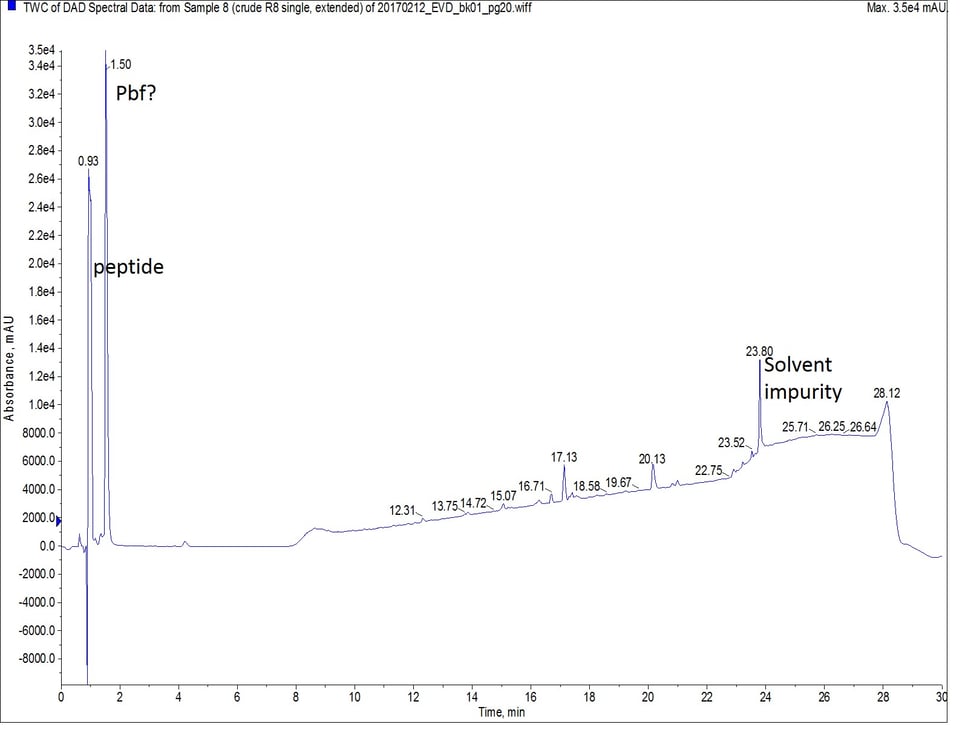
Figure 3: Crude analytical UV HPLC spectrum of octa-Arg synthesized with single, extended couplings. There is no evidence of the peptide eluting later in the gradient. The mass spectrum of the predominant peptide peak (r.t. = 0.93 min) is the same as observed in Figure 2.
Finally, I synthesized octa-Arg using a double coupling strategy for the entire peptide sequence. I kept the scale, coupling reagents, and equivalents constant with the previous two syntheses, and each coupling reaction was allowed to proceed at 75°C for 5 minutes. While this synthesis yields the purest crude peptide, Figure 4, it also consumed double the amount of amino acid as the other two synthetic efforts.
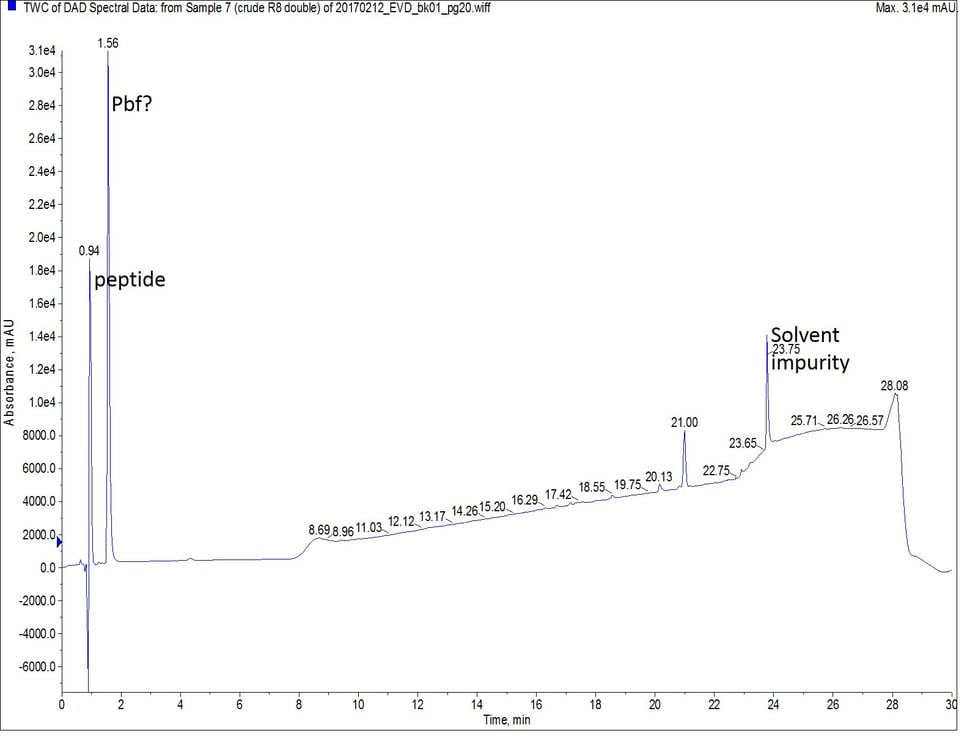
Figure 4: Crude analytical UV HPLC spectrum of octa-Arg synthesized with double couplings. There is no evidence of the peptide eluting later in the gradient. The mass spectrum of the predominant peptide peak (r.t. = 0.94 min) is the same as observed in Figure 2.
There is one glaring item I haven’t discussed at this point, and that is the identity of the significant peak that elutes at approximately 1.5 minutes. The peak contains no mass signature below 500 Da, the molecular weight cutoff I typically use for my mass analysis of peptides. But it’s strong UV signature causes me to second guess it’s identity. My best guess at this point is that the peak contains the Pbf protecting group. I came to this conclusion based on two critical facts: 1) there is an 8-fold excess of Pbf for every peptide molecule present in the sample and 2) the dihyrdobenzofuran core of Pbf absorbs strongly in the UV. I’ll definitely be repeating this analysis with a lower mass range to confirm these conclusions.
Well, I had hoped that this experiment would be a little more interesting. I shouldn’t be complaining that my “challenging” syntheses were successful, despite my efforts to help them fail. After staring at the UV spectra for a while it slowly becomes clear that the double coupling strategy seems to yield a cleaner crude peptide sample. It is most interesting to me that the peptide synthesized using extended single couplings also had the lowest crude purity. Even though the differences aren’t wildly obvious with this series of peptides, I still recommend double coupling arginine residues during your peptide synthesis, especially if the arginine is adjacent to a bulky amino acid.
Want to learn how to reduce your time spent performing peptide materials preparation - synthesizing peptides for use in real experiments?