 Organic Workflow
Organic Workflow Peptide Workflow
Peptide Workflow Scale-Up Flash Purification
Scale-Up Flash Purification  Sample Preparation
Sample Preparation Biomolecule Purification
Biomolecule Purification Oligo synthesis
Oligo synthesis Scavengers and Reagents
Scavengers and Reagents Service & Support
Service & Support Accessories & Spare parts
Accessories & Spare parts Investors
Investors Reports & News
Reports & News The Share
The Share Corporate Governance
Corporate Governance Calendar
Calendar Sustainability
Sustainability Our Offering
Our Offering Our History
Our History Our Locations
Our Locations Leadership
LeadershipWith most chromatographic purifications, only two solvents are needed to adequately separate compounds from each other. Unfortunately, there are instances where the separation is either poor or cannot be accomplished with “normal” elution conditions such as those with ionic or very polar organic molecules.
In this post I offer some solutions to this issue.
Ionizable compounds are typically water miscible and are best separated/purified using reversed-phase methods. The challenge with these water-soluble compounds is getting them to retain on hydrophobic (i.e. C18) media and elute in a tight band, the goal if you seek to purify them. To ensure both of these goals are met, pH modifiers or buffers are typically needed.
Why? Because ionic and ionizable compounds strive to achieve equilibria in solution (remember pKa, pKb, etc. from your undergraduate college days?). These compounds have their counter ions partially "attached" and partially disassociated from the parent compound. Because these molecules exist in different states of equilibria, chromatography becomes challenging. The ionized form is more polar and tends to elute early while the non-ionized for is retained - this results in tailing or fronting peaks, Figure 1.
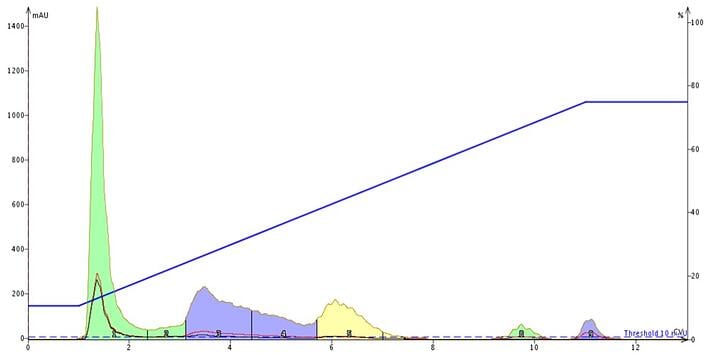 Figure 1. Chromatogram of ionic organic compounds - tartrazine, allura red, brilliant blue FCF, and erythrosine (all food colors) - using a water/methanol gradient. The highly ionic and dissociative nature of the compounds minimizes their ability to partition with the C18 due to high polarity. To remedy this issue you need to either adjust pH to force the equilibrium into one state or the other or add a buffer to force counter ion association.
Figure 1. Chromatogram of ionic organic compounds - tartrazine, allura red, brilliant blue FCF, and erythrosine (all food colors) - using a water/methanol gradient. The highly ionic and dissociative nature of the compounds minimizes their ability to partition with the C18 due to high polarity. To remedy this issue you need to either adjust pH to force the equilibrium into one state or the other or add a buffer to force counter ion association.
For acidic and basic compounds, I like to follow the 2 pH rule, if possible. The 2 pH rule states that to protonate an acid, the mobile phase pH must be 2 pH units below the compound’s pKa. Likewise, to force organic amines into the free-base form the mobile phase pH should be 2 pH units above the compound’s pKa. The benefits in both cases is that the compounds are rendered more hydrophobic and therefore have increased retention on the reversed-phase column, which usually provides a better separation with improved loading capacity and peak shape (increased fraction concentration). Examples of both situations follow.
A. Acidic compounds are best retained and separated when the mobile phase is acidic. I use a volatile acid at the 0.1% level in both the weak and strong eluting solvents. Why volatile acids? Because they are easier to evaporate when you need to recover your compound from the collection vessel.
Suggested acids include...
i. Formic
ii. Acetic
iii. Trifluoracetic (TFA)
In Figure 2 we show how reversed-phase chromatography with the addition of an organic acid (TFA) helps to retain and separate three small organic acids including 3-nitrobenzoic acid (pKa 3.47) , nicotinic acid (pKa 4.85), and homovanillic acid (pKa 3.74). With a low pH mobile phase like the one used here, these acids are protonated and made more hydrophobic which enhances retention and increases the separation, making for successful flash purification.
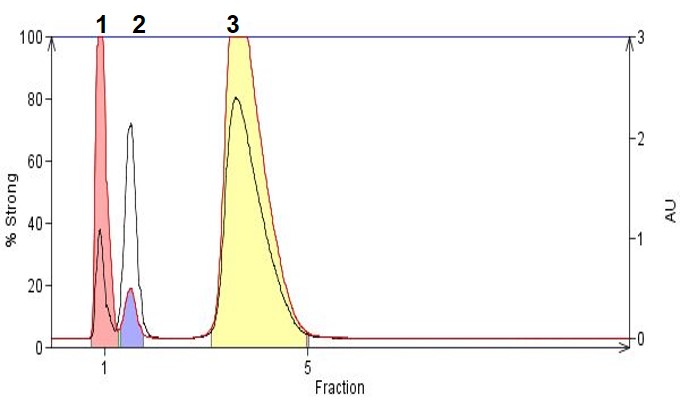
Figure 2. Organic acid purification by reversed-phase using a mobile phase of acetonitrile/water/TFA (60:40:0.05). Peak 1 is 3-nitrobenzoic acid, Peak 2 is nicotinic acid, Peak 3 is homovanillic acid.
B. Similarly, basic compounds are best retained and separated when the mobile phase pH is alkaline. I like to use volatile bases at the 0.1% level in both the weak and strong solvents, examples of which include...
i. Ammonium hydroxide
ii. Diethylamine (DEA)
iii. Triethylamine (TEA)
In Figure 3 we show an example where three organic amines, 4-dimethylamino antipyrine (pKa 9.6), N-phenylbenzylamine (pKa 3.5), and amitriptyline (pKa 9.4) are separated using a gradient incorporating TEA as the base. As with the example above with organic acids, adjusting the mobile phase pH 2 units beyond the compounds' pKa, in this case to ~pH 11, improves hydrophobicity and retention. This, in turn, improves the separation and simplifies fraction recovery (less water, more organic solvent in the collected fractions).
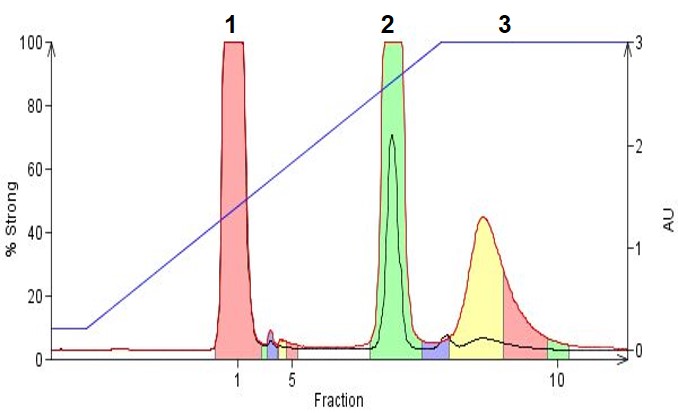
Figure 3. Organic amines purified using reversed-phase gradient flash chromatography. Solvent used were acetonitrile, water and 0.1% TEA. Peak 1 is dimethylamino antipyrene, Peak 2 is N-phenylbenzylamine, and Peak 3 is amitriptyline.
For charged compounds / organic salts you can use the above strategies but if your compound or compounds are strong acids, strong bases, or zwitterionic (as with the food colors example in Figure 1) then adding a volatile buffer can do wonders for the separation. In these instances, I like to choose from the following…
1. Ammonium formate (10 mM)
2. Ammonium acetate (10 mM)
Continuing with the food colors example, we show the chromatographic separation improvement when a volatile buffer is added, Figure 4. The buffer in this case was ammonium acetate, pH 7. By adding a volatile organic buffer the sample's charged sites are "neutralized" with counter ions.
Since the buffer concentration is greater than that of the sample components, the sample's equilibrium is forced into this paired-ion, neutralized state which makes the compounds more lipophilic and therefore, more retentive. Pairing the ionic sites with a counter ion also minimizes chemical ionization which can impact peak shape / band broadening seen below.
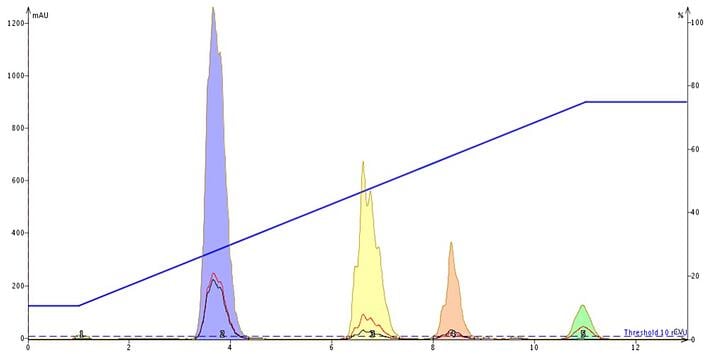 Figure 4. Adding an ammonium acetate buffer, pH 7, to the solvent system makes the ionic compounds more lipophilic and more retentive on reversed-phase media. Peak shape is improved which minimizes fraction volume.
Figure 4. Adding an ammonium acetate buffer, pH 7, to the solvent system makes the ionic compounds more lipophilic and more retentive on reversed-phase media. Peak shape is improved which minimizes fraction volume.
By adding a buffer to the mobile phase you also increase its ionic strength. Added ions increase mobile phase polarity which can be helpful in improving very polar compound retention and separation as seen with the purification of a DMSO solution containing uracil and caffeine, Figure 5. Increasing the mobile phase ionic strength, and therefore polarity, allows uracil to better partition with the C18 and be at least partially separated from DMSO; caffeine's peak shape also improves with ionic strength.
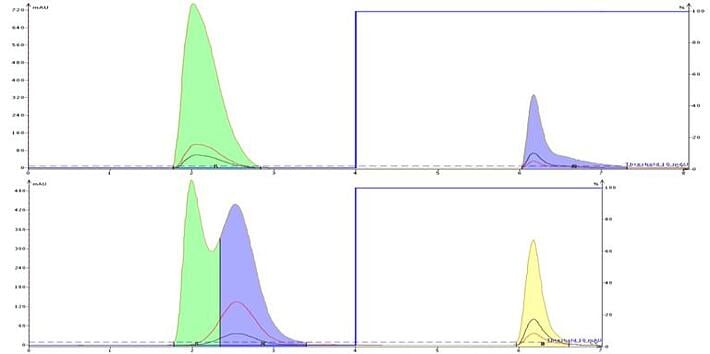 Figure 5. Non-ionic compounds also can benefit from buffers who's ion make the solvents more polar. In the top chromatogram, uracil co-elutes with the injection solvent, DMSO, while caffeine is retained but shows some tailing. When buffer is added, uracil's retention is improved and caffeine elutes in a much tighter band.
Figure 5. Non-ionic compounds also can benefit from buffers who's ion make the solvents more polar. In the top chromatogram, uracil co-elutes with the injection solvent, DMSO, while caffeine is retained but shows some tailing. When buffer is added, uracil's retention is improved and caffeine elutes in a much tighter band.
So, to create purification methods best suited to your sample's ionic or ionizable chemistry, choose the right solvent conditions (add acid, base, or buffer) to maximize separation efficiency and loading while minimizing fraction volume.
For more information on flash method development and optimization, download our whitepaper - Successful Flash Chromatography.